Needless to say, the new EU Regulation on medical devices (MDR 2017/745) and the EU Regulation on in-vitro diagnostics (IVDR 2017/746) are challenging for all involved stakeholders. Whether authorities, notified bodies, manufacturers, distributors, importers and other subcontractors/suppliers involved – the new regulations and their increased requirements are burdensome and will be a major game changer.
The European Commission published some high level transition guidances for medical device and IVD companies which are a good basis to plan a stepwise preparation. But everybody moving into deeper details will realize it as a Herculean task demanding both a solid strategy as well as respective resources.
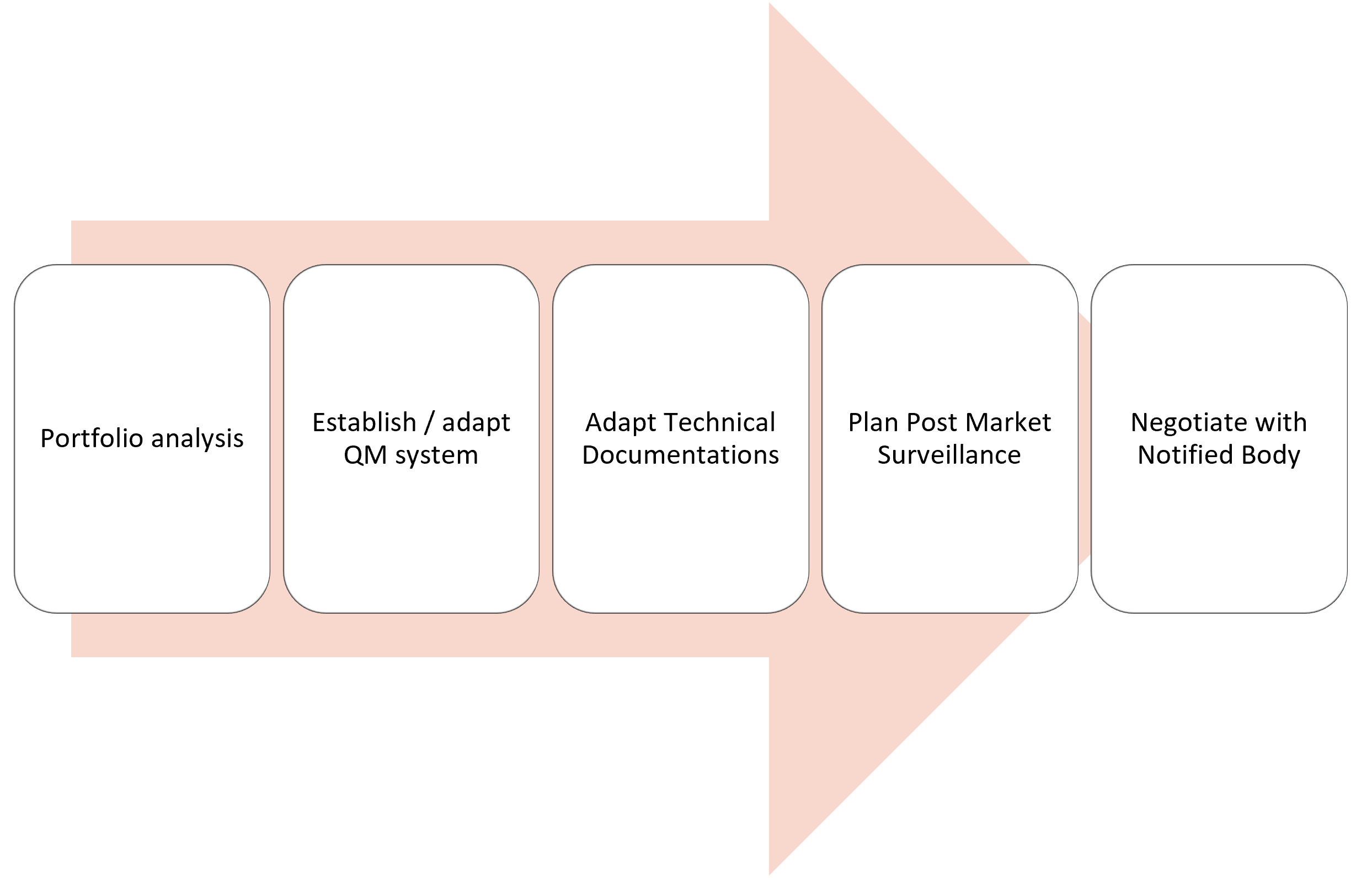
CE plus can help you in both regards: define a custom-fit transition strategy and provide substantial hands-on support. Our MDR/IVDR transition services include: